

Nature: New Progress in C- H Activation!
2023-08-01
FGs fundamentally affect the properties and functions of organic molecules, making the transformation of introducing, removing or interconverting FGs of high importance in organic chemistry and its related fields. In addition to regular FG interconversion reactions that primarily alter the diversity of FGs, new transformations directed to the introduction of FGs at remote C- H sites, such as C- H functionalization and migration functionalization reactions, have attracted significant attention.


First Author:Ken Chen
Correspondence Author:Yan Xu (Xu Yan)
Address: Peking University
Article link: https://doi.org/10.1038/s41586-023-06347-3
research background
1. FGs fundamentally affect the properties and functions of organic molecules, making the transformation of introducing, removing or interconverting FGs of high importance in organic chemistry and its related fields. In addition to regular FG interconversion reactions that primarily alter the diversity of FGs, new transformations directed to the introduction of FGs at remote C- H sites, such as C- H functionalization and migration functionalization reactions, have attracted significant attention.
2. But in the network of chemical connections and disconnections, a unique classFG manipulation transformation, I .e. direct repositioning of FG to an unactivated or even traditionally inaccessible C- H site, without introducing any concomitant changes to the molecule, is still rarely explored (FIG. 1a). This type of direct FG translocation reaction contrasts and/or complements the reaction types described above, exhibiting molecular editing properties and an ideal atom economy of 100 percent. In addition, exploring the challenging C- H chemistry space may also become possible.
3. To date, in addition to the intra-chain isomerization of olefins and alkynes, this type of directFG translocation reactions have hardly been reported before. Dispersed examples can be found in biocatalysis using substrate-specific mutases, Pd-catalyzed aromatic ester "dancing" through aryl-type intermediates, or others, but all of them are of limited generality.
4. Introduction, removal, or manipulation of functional groupsChemical transformations of (FG) are ubiquitous in synthetic chemistry. Unlike the traditional FG interconversion reaction, which exchanges one functional group for another, species transformations that only change the FG position have been much less studied.
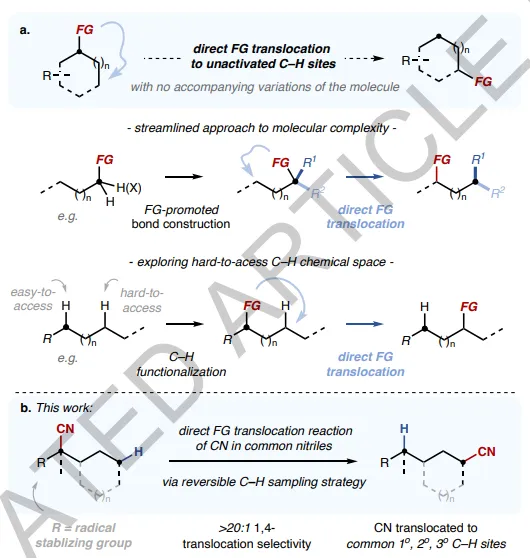
Figure1 Direct translocation of functional groups to unactivated C- H sites
research content
1. In this paper, through photocatalysis, reversibleC- H sampling, the authors report FG metathesis reactions of the cyano (CN) group in common nitriles, allowing direct position exchange between CN and the unactivated C- H bond. This reaction shows a high fidelity of 1,4-CN translocation, which is generally contrary to the site selectivity inherent in traditional CH functionalization.
2. This paper also reports the direct cross-ring of the circulatory systemThe CN translocation provides access to valuable structures, while obtaining these structures by other methods is non-trivial. Taking advantage of the synthetic versatility of CN and the key CN translocation step, the authors demonstrate the concise synthesis of biologically active molecular building blocks.
3. in addition,The combination of C- H cyanation and CN metathesis allows to obtain unconventional C- H derivatives. Overall, the reported reaction represents a method to achieve a site-selective C- H conversion reaction without a site-selective C- H cleavage step.
graphic analysis
With respect to reactive design, the authors first assume thatIntramolecular radical addition by CN followed by β-fragmentation of the generated imino radical can be used to migrate CN (FIG. 2a, steps B and C). With the catalytic "cycle" nearly closed, key challenges regarding site selectivity need to be addressed. Site-selective HAAs between similar and unactivated C- H bonds are challenging. However, the authors propose that it may be feasible to better control the overall site selectivity of the translocation reaction by employing a reversible C- H sampling strategy. This strategy will involve reversible and non-specific cleavage of a series of similar C- H bonds, followed by selective functionalization of the preferred C- H sites for subsequent steps (Figure 2b). Here, this would require establishing net reversibility in the less selective HAT steps between different C- H sites (I. e., steps A and E) by cooperative HAA/HAD catalysis, and combining this reversibility with the more site-sensitive radical addition step to differentiate the generated carbon radicals (step B). In addition, a radical stabilizing substituent at the CN α position is important for facilitating the translocation. The reversible C- H sampling strategy proposes an unconventional paradigm that can achieve site-selective C- H conversion reactions without the need for a site-selective C- H cleavage step, which provides a potentially useful framework for functionalizing more diverse substrates containing multiple types of similar C- H bonds. Nevertheless, 2-methyl-2-pentylmalononitrile (1) was selected as a model substrate to verify the author's hypothesis (Figure 2c).
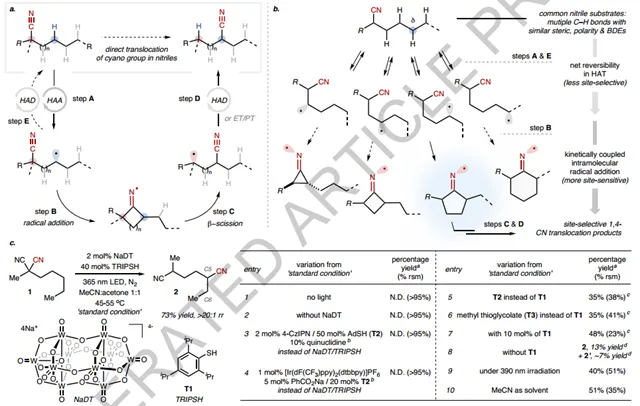
Figure2 Design and optimization of CN translocation reaction
Next check the substrate range (Fig.3). First, the high regioselectivity identified by the authors is ubiquitous (> 20:1 in all cases) and is not affected by alkyl length (e. g., in 13, 15, 17, and 19). For example, even for a sample containing 8 similar-CH2Substrate 19 of the-unit, the exclusive C4 selectivity also remained unchanged, although lower conversions were observed. In some cases, the fidelity of the 1,4-translocation was found to exceed the typical site preferences in traditional HAT-based C- H functionalization. Notable examples include substrates 96 29(1 ° C- H over 2 °),31 (hindered 2 ° C- H over 3o), 33(2 ° C- H over ether α C- H), and 35(2 ° C- H over benzyl). A variety of malononitrile can be converted to valuable 1,4-dinitriles with different functionalization modes (e. g., 22 and 97). "Mono"-nitrile substrates are compatible under slightly modified conditions as long as they possess a radical stable α-substituent, such as α-cyanoester (37), ketone (51), arene (39-41, 53) and heteroarene (47). Alpha-cyanated diesters (55) and amino esters (49, 101-104) were also tested, resulting in novel derivatives with precise introduction of remote CN groups. Translocation of CNs to tertiary (99), secondary or even inactive primary (30) C- H sites is possible. Functional groups-including alkyl chlorides (11), aryl bromides (26) and boronic esters (10), tertiary alcohols (12), phosphinates (50), thiophenes (108) and ketones (52)-are tolerated. Consistent with the mechanistic hypothesis, the diastereoselectivities of most linear substrates are below 2:1 due to uncontrolled C- H-forming steps. However, products whose diastereomeric proportions are determined in the radical addition step may exhibit high diastereoselectivity. It is worth noting that in almost all nitriles examined, whether through 1-electron or 2-electron processes, the CN group is repositioned to a position that is not the most accessible C- H site in nature, whether through 1 or 2-electron processes. This emphasizes the potential of this method in accessing difficult-to-manipulate C(sp3 )-H bond data. Finally, the 100 allyl-containing C- H bonds are incompatible, resulting in a complex reaction mixture.
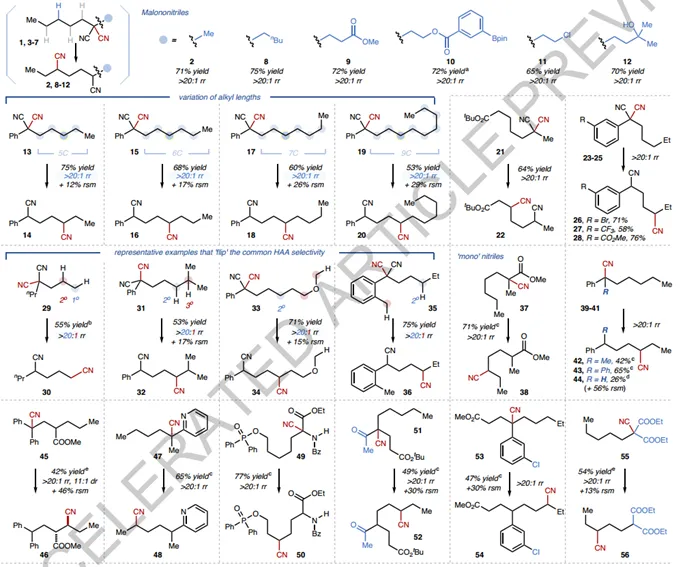
Figure3 Substrate range of linear nitriles
Next, the authors examine the direct span ringCN translocation, where bridged (and typically strained) bicyclic intermediates are involved (Figure 4). The addition of carbon radicals to CN to form such intermediates can be kinetically challenging. The ability of the photocatalytic CN metathesis system to (re) generate the desired carbon radicals from C- H bonds counteracts the risk of premature carbon radical quenching compared to classical radical generation schemes using halides or alkenes. In fact, transcyclic CN translocations occur smoothly in different structures. Various six-membered rings (67, 70), seven-membered rings (58, 61, 64, 76) and eight-membered rings (73) were found to be suitable, with the CN being placed on the cyclic backbone or side chain. Excellent 1,4-translocation selectivity was observed in all cases, and an additional 17:1 γ-selectivity was obtained using α-cyanocycloheptanone 64, which contains two potential 1,4-translocation destinations. It is worth noting that taking advantage of the synthetic versatility of CN, the synthesis of all cyclic nitrile substrates is completed in 1-2 steps from the raw material chemicals. This, combined with the CN translocation method, provides a direct route to the construction of a variety of polysubstituted carbocycles, which are not simple to construct. For example, previously laborious synthesis of the same functionalization pattern as in 80 can now be accomplished by a concise two-step sequence of 1-benzocycloketone (78). In another example, the site of 82 and the diastereoselective CN translocation enables the precise introduction of CN in the framework of 9 C- H bonds, which is otherwise difficult to achieve, and provides a novel building block (83) with unique three-dimensional properties.
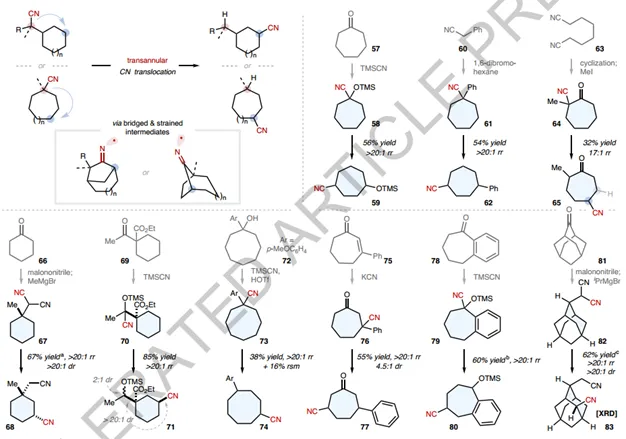
Figure4 Direct transring CN translocation
The authors also conducted other studies to demonstrate the potential utility of the approach (Fig.5). First, the "C-H cyanide-CN translocation" strategy was demonstrated by using a selective RARβ2 agonist (AC 55649) and γ-alkylated tetrahydropyran (87) as substrates. By first introducing CN at C- H sites that are susceptible to functionalization, and then selective CN translocation, accurate access to novel cyanated derivatives at unconventional C- H sites was achieved (Figure 5a). Secondly, the method can be used for the late modification of some complex scaffolds, such as derivatives of steroids (91), peptides (92) and bioactive molecules (90) (Figure 5b). Neither multifunctional coupling partners nor laborious routes are required, and the products can be further diversified into simple conversion derivatives of amides (112), ketones (114), aldehydes (115), protected amines (113) and 137 diaminopyrimidines (116)CN.
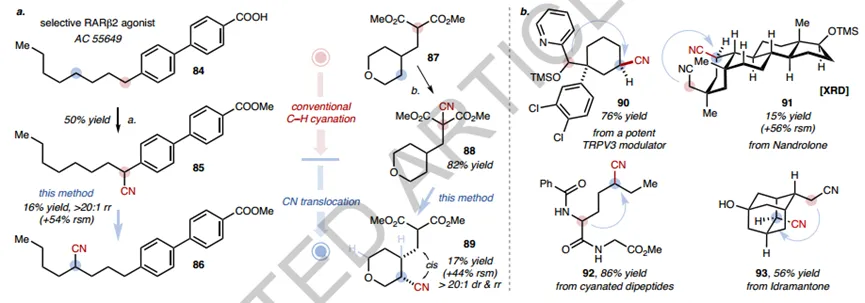
Figure5 Application of direct CN translocation reaction
in addition,The synthetic versatility of CN and the CN translocation reaction can be combined to speed up the synthesis of valuable building blocks. For example, the diester 120, a key intermediate for HBV inhibitors, was previously prepared in 7 steps; here, the 120 step was obtained from methyl 2-cyanoacetate in only 3 steps by employing CN-promoted nucleophilic substitution, CN metathesis, and CN conversion to a ketoester. The synthesis of precursors of estrogen receptor-β agonists is also done with the help of CN translocation, again providing a simplified alternative to the 7-step literature procedure. The author's laboratory is exploring other FG translocation reactions and utilizing reversible C- H sampling strategies for further studies.

Artikel sebelumnya
Berikutnya
Informasi terkait






Hubungi kami
Situs web: Website
Alamat: No. 66 jalan selatan Xinyuan, distrik Haicang, Kota Xiamen, Provinsi Fujian, Tiongkok

Kode QR akun resmi